Cystic fibrosis
This assignment was to explain the “Genetic, Molecular and Physiological Basis of the Respiratory Problems Associated with Cystic Fibrosis” in 400 words. A full bibliography of sources is provided at the end. Click on figures and diagrams to enlarge them.
Please remember that this is merely a medical school assignment, and does not constitute medical advice.
The Genetic, Molecular and Physiological Basis of the Respiratory Problems Associated with Cystic Fibrosis
Cystic fibrosis (CF) is caused by the lack of a normal cystic fibrosis transmembrane conductance regulator (CFTR) protein (fig 1). Sufferers have only ‘faulty’ CFTR proteins, which arise from a genetic mutation on the long arm of chromosome 7. The condition is recessive, meaning that an affected gene must be inherited from both parents in order for the child to exhibit the condition. The disease affects around 1 in 2415 live births in the UK, implying a gene frequency of about 1 in 25 (Dodge et al, 1997). A deletion mutation at position 508 in the amino acid sequence (known as ∆F508) is the most common mutation, causing 88% of CF cases (Kingston, 2002). This mutation causes the protein resulting from the translation of the genetic material to be incorrectly structured. In CF, the secondary and tertiary structures of the CFTR protein are incorrect, meaning that it is incorrectly ‘packaged’ by the Golgi apparatus, and so does not take up the normal position on the cell surface membrane.
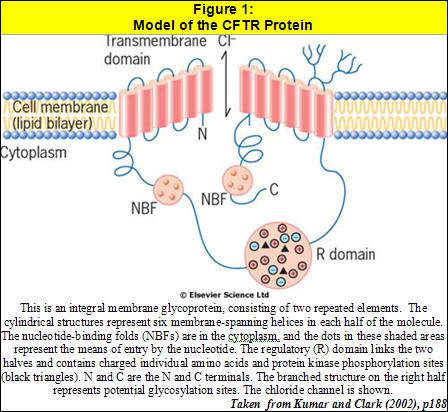
In an unaffected individual, CFTR functions as a chloride ion channel in epithelial cells, opening in response to increased levels of intracellular cyclic adenosine monophosphate (cAMP). Therefore, a defective CFTR will cause abnormal chloride ion transport, leading to abnormal conductance of salt and water in epithelial cells. The respiratory tract is affected since there is a decreased excretion of chloride into the lumen of the airway, and a threefold increase in the reabsorption of sodium into the epithelial cells. The reduced amount of salt in the lumen results in a change to the normal osmotic pressures: less water than normal will be drawn into the lumen by osmosis, resulting in increased viscosity and tenacity of the mucus in the airway (Kumar and Clark, 2002).
The abnormal mucus provides ideal conditions for bacteria not usually found in the respiratory tract, such as pseudomonas. Cell destruction through infection in the airways causes cell debris to appear in the mucus, increasing its viscosity. Since DNA is a large molecule, this component of the cell debris drastically increases the viscosity of the mucus (Khan et al, 1995).
The modified mucus and infections cause a number of problems for the patient, particularly since many infections cause inflammation of the bronchioles – a combination of bronchial inflammation and thick mucus can cause severe breathing difficulties. Commonly documented respiratory symptoms of cystic fibrosis are breathlessness, sinusitis, nasal polyps and coughing up thick sputum. Haemoptysis may also occur, especially if the lungs are particularly badly infected (Kumar and Clark, 2002).
Bibliography
Abbott, J. (2003), Coping with cystic fibrosis. Journal of the Royal Society of Medicine, 96(Suppl. 43):42-50
Dodge, J.A., Morison, S., Lewis, P.A., Coles, E.C., Geddes, D., Russell, G., Littlewood, J.M. and Scott, M.T. (1997), Incidence, population, and survival of cystic fibrosis in the UK, 1968-95. Archives of Disease in Childhood, 77(6):493-6
Doull, I (2001), Recent advances in cystic fibrosis. Archives of Disease in Childhood, 85(1):62-6
Hunter, V. (2003), The daily grind and how to stay sane as a mother of two children with cystic fibrosis. Journal of the Royal Society of Medicine, 96(Suppl. 43):51-56
Kent, G., Iles, R., Bear, C.E., Huan, L.J., Griesenbach, U., McKerlie, C., Frndova, H., Ackerley, C., Gosselin, D., Radzioch, D., O’Brodovich, H., Tsui, L.C., Buchwald, M. and Tanswell, A.K. (1997), Lung Disease in Mice with Cystic Fibrosis. Journal of Clinical Investigation, 100(12):3060-9
Kettler, L.J., Sawyer, S.M., Winefield, H.R. and Greville, H.W. (2002), Determinants of adherence in adults with cystic fibrosis. Thorax, 57(5):459-64
Khan, T.Z., Wagener, J.S., Bost, T., Martinez, J., Accurso, F.Z. and Riches, D.W. (1995), Early pulmonary inflammation in infants with cystic fibrosis. American Journal of Respiratory and Critical Care Medicine, 151(4):1075-82
Kingston, H.M. (2002) ABC of Clinical Genetics, 3rd ed. London: BMJ Books » Buy Now
Kumar, P. and Clark, M. (2002) Clinical Medicine, 5th ed. London: WB Saunders » Buy Now
Lodish, H., Berk, A., Matsudaira, P., Kaiser, C.A., Krieger, M., Scott, M.P., Zipursky, S.L. and Darnell, J. (2003) Molecular Cell Biology, 5th ed. New York: WH Freeman » Buy Now
Lowton, K. and Gabe, J. (2003) Life on a slippery slope: perceptions of health in adults with cystic fibrosis. Sociology of Health and Illness, 25(4):289-319
Quinton, P.M. (1999) Physiological Basis of Cystic Fibrosis: A Historical Perspective. Physiological Reviews, 79(1):S3-22